Risk‑based classification: your first decision point
The very first question to answer is whether your product is a medical device. Most regulators follow the International Medical Device Regulators Forum’s risk‑based framework: software used solely to store or display data may not be considered a device; once it analyzes or influences medical decisions, it almost certainly is. Classification determines the regulatory pathway:
- Low‑risk devices (Class I): Administrative or wellbeing apps, often self‑declared in Europe and exempt from premarket review in the U.S.
- Moderate‑risk devices (Class II): Most clinical decision‑support tools and non‑critical AI diagnostics. These require a Notified Body review in the EU (Class IIa/IIb) and a 510(k) or De Novo submission in the U.S.
- High‑risk devices (Class III): Software that diagnoses or treats life‑threatening conditions or controls implants. These require rigorous clinical evidence and the longest review times. A structured decision tree helps. Answering these questions narrows the class and directs you to the correct route.
Is it a Medical Device?
1. Does the software have a medical purpose (diagnosis, treatment, prevention, monitoring, or alleviation of disease/injury)? o No → Not a medical device; may still require data protection compliance (e.g., GDPR, HIPAA). o Yes → proceed.
2. Does the software drive or control a medical device or provide therapy/diagnosis?
o Yes → regulated as part of the parent device; follow the highest class. o No → proceed.
3. What is the significance of the information to the healthcare decision? (align with IMDRF SaMD framework and EU MDR Rule 11【15†L179-L187】) o Inform clinical management (e.g., documentation, administrative support) → Risk Category I → Class I (EU/UK) or Class I/II (US) → Self‑certification or exempt. o Drive clinical management (e.g., calculates drug dosage) or diagnose/treat non‑serious condition → Risk Category II–III → Class IIa/IIb (EU), Class II (US). Requires NB or 510(k)/De Novo review. o Diagnose/treat serious or critical condition or failure could result in death/serious deterioration → Risk Category IV → Class III (EU/US/JP). Needs PMA or EU Class III NB design examination with a clinical trial.
4. Is the software adaptive AI/ML?
o If yes, check if the regulator has an adaptive algorithm framework. US FDA allows a PCCP to pre‑authorise changes【27†L29-L36】; Japan’s PMDA uses IDATEN for post‑approval changes. EU currently requires re‑assessment for significant software changes, with the AI Act to introduce risk management and transparency obligations【19†L105-L113】.
Regulatory timelines across regions
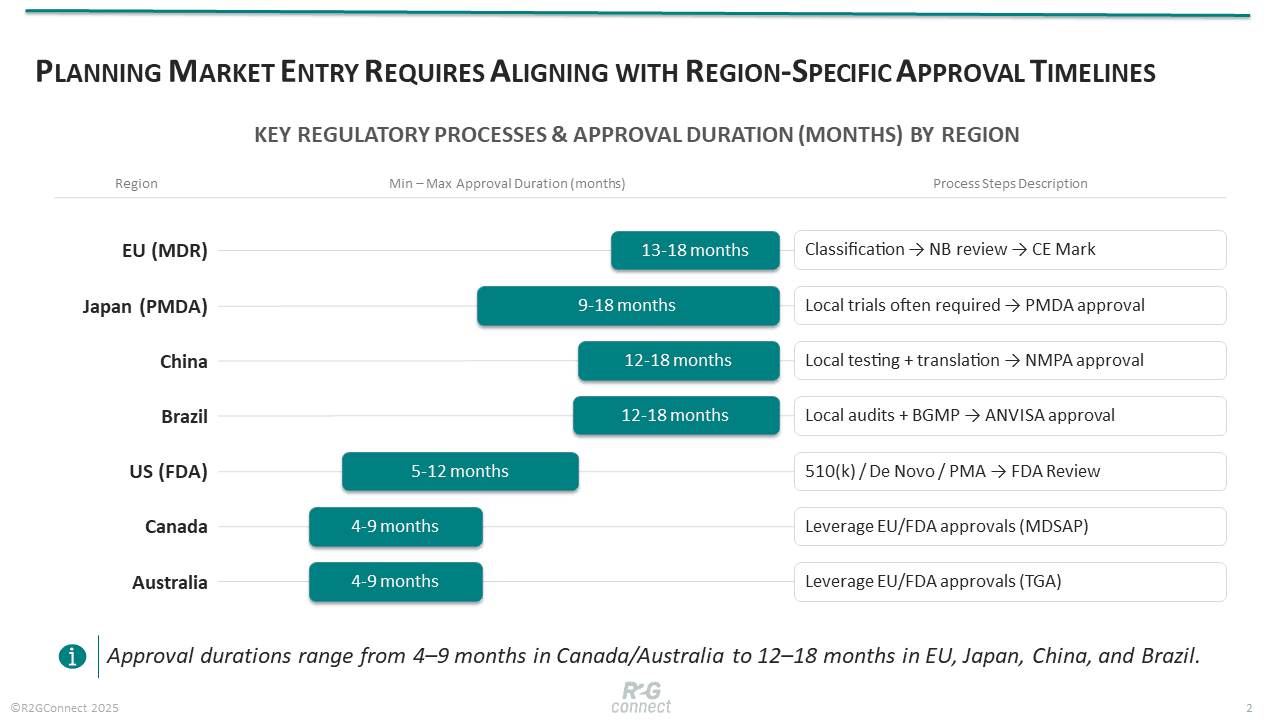
Approval durations vary by region and risk class. In the EU, MDR’s conformity assessment can take 9–24 months, with an industry survey reporting average lead times of 13–18 months; planning should factor in broad timeframes of 6–14 months. These long queues stem from the limited number of designated Notified Bodies and the complexity of the new MDR requirements. The UK still accepts CE marks until at least 2027–28, but a new UKCA regime is imminent.
The U.S. offers comparatively faster pathways: a traditional 510(k) review nominally targets 90 days, though with interactive review cycles it often runs 5–7 months; De Novo classifications can take 8–12 months. The FDA is also adopting modern approaches, such as Predetermined Change Control Plans for AI/ML devices and cybersecurity mandates requiring threat models and software bills of materials. Japan’s PMDA approvals average 9–18 months for Class III devices, while Canada and Australia often leverage foreign approvals to shorten review to 4–9 months. Emerging markets like China and Brazil impose local testing and longer documentation cycles, often extending timelines to a year or more.
Step‑by‑step process, phased for start‑ups
MVP / Concept phase (0–6 months) 1. Classify and scope. Determine if your product qualifies as a medical device and which class it falls into using the decision tree above. This shapes everything that follows. 2. Map regulatory strategy. Identify target markets early and determine whether to pursue approval in the U.S., EU or other jurisdictions. Understanding the differences between MDR, FDA and other pathways helps define a clear market‑entry sequence. 3. Build a foundational QMS. Even before clinical work, create an ISO 13485‑compliant quality management system (QMS) covering design controls, risk management and document control. Starting early allows you to integrate regulatory requirements into your development process from day one. 4. Plan your software lifecycle and risk management. Implement IEC 62304 processes (requirements, design, verification, validation) and ISO 14971 risk analysis. For AI software, begin documenting datasets, training methods, and bias mitigation.
Pilot / Clinical validation phase (6–18 months) 1. Pre‑submission engagement. In the U.S., request a Q‑submission meeting with the FDA to get feedback on your classification, test plans, and evidence requirements. In Europe, consult your Notified Body about classification and documentation expectations. 2. Design and conduct studies. Compile existing clinical evidence or perform new trials. Under MDR, even Class IIa software requires a clinical evaluation report summarising published data and post‑market experience. High‑risk software may need prospective clinical investigations and multi‑site studies coordinated across regions. 3. Fill your technical file. Maintain a living document of device description, software architecture, risk management, verification reports, usability studies, clinical evidence, and cybersecurity plans. Periodically audit your documentation against MDR or FDA requirements before submission. 4. Establish local representation. If targeting the EU or other non‑domestic markets, appoint a legal representative or importer and plan translations. Data localisation rules (e.g., in China and Saudi Arabia) can add complexity.
Scale / Submission phase (18–36 months) 1. Finalise submissions. Prepare your 510(k), De Novo, or PMA application for the FDA and your CE/UKCA dossier for the EU/UK. Include your QMS certificates, GSPR checklist, clinical data, and cybersecurity documentation. For AI, include the Predetermined Change Control Plan if applicable. 2. Respond to queries. Regulators will likely issue deficiency letters; respond promptly with clarifications and additional data. Delays often occur if the technical file lacks a complete risk analysis or evidence. 3. Parallel pathways. Consider concurrent submissions: some firms file in the U.S. and Canada simultaneously, then Europe, or use EU approval to leverage faster reviews in Australia and Singapore. The sequence depends on market priorities and reimbursement opportunities. 4. Plan for post‑approval. Implement a post‑market surveillance (PMS) system: track adverse events, monitor cybersecurity, and schedule periodic safety update reports (PSUR) in the EU. For AI/ML tools, prepare to submit algorithm updates under the PCCP or via new submissions.
Post‑market / Continuous improvement phase 1. Monitor performance and safety. Collect real‑world evidence, track user complaints, and update risk analyses. EU MDR requires annual PSURs for high‑risk devices; the FDA expects prompt adverse‑event reporting. 2. Manage software updates. For SaaS products, updates may trigger re‑submission. Use pre‑agreed PCCP frameworks (FDA) or consult Notified Bodies to determine if an update is “significant” under MDR. 3. Maintain QMS and audits. Expect annual surveillance audits in Europe and MDSAP jurisdictions. Keep QMS procedures updated to reflect process changes. 4. Expand market access. Once initial approvals are in place, plan entry into new regions. Consider local registration requirements (e.g., Japanese language labeling, Brazilian BGMP audits). Each new market may require additional timelines, typically 6–12 months.
Practical takeaways for founders
-
Classify correctly. Invest time in understanding the risk‑based classification in your target markets. Misclassification leads to under‑ or over‑documentation and delays. A free consultation like BAYOOCARE’s roadmap can be invaluable.
-
Start your QMS early. Even if you’re pre‑clinical, set up an ISO 13485‑compliant QMS. BayooMED’s fast‑track program is a cost‑effective way to build the backbone for your technical file and risk management.
-
Audit your documentation. Use gap analyses from experts like CEED | Charité to identify missing elements before a Notified Body or FDA reviewer does. It’s cheaper to fix gaps early than after a failed submission.
-
Plan for evidence. Budget for clinical studies and engage regulatory experts (e.g., CoLAB TRIALS) to design them. Early regulatory feedback can drastically reduce the need for additional trials.
-
Allocate for post‑market. Monitor safety, manage cybersecurity updates, and prepare periodic reports. Don’t view approval as the finish line; regulators watch performance throughout the product’s life.
-
Strategize market entry. Consider the order of approvals. For many, the U.S. first followed by the EU and Asia‑Pacific, may maximize time‑to‑market and investor confidence. Others may leverage the EU’s DiGA or PECAN reimbursement for early revenue. Always factor in local language, representation, and privacy laws.
-
Develop a robust AI and cyber strategy. Emerging regulations like the EU AI Act and cyber‑resilience requirements demand careful planning. TÜVIT’s EU AI Act Briefing & AI Risk Navigator is a free consultation that helps AI/ML developers understand high‑risk requirements, risk management, and documentation expectations, preventing hidden compliance costs.
-
Explore reimbursement pathways. In Germany, the DiPA reimbursement framework can provide early revenue for digital nursing‑care applications. TÜVIT’s DiPA Reimbursement Consultation outlines how to qualify under this pathway, helping you offset regulatory expenses.
By understanding the timelines, managing each step thoughtfully, and tapping into trusted services like those on R2GConnect, healthtech founders can chart a clearer path through the regulatory maze and bring transformative digital health solutions to patients faster and more safely.